Welcome

Welcome to the Goerigk Research Group website. We are part of the Melbourne Centre for Theoretical and Computational Chemistry at the School of Chemistry, The University of Melbourne, Australia. Our research revolves around the exciting field of Quantum Chemistry, which is the description of electrons in chemical systems by evoking the laws of Quantum Mechanics. Our interests comprise both the development of new quantum-chemical methods, as well as applications to Organic-, Inorganic-, Physico- or Biochemical prob![]() lems. We have established a range of national and international collaborations with experimental and other theoretical groups, and our research is supported by various local, national and international funding schemes. Please feel free to explore our website through the links provided in the right-hand sidebar (or at the bottom of the page, depending on your device).
lems. We have established a range of national and international collaborations with experimental and other theoretical groups, and our research is supported by various local, national and international funding schemes. Please feel free to explore our website through the links provided in the right-hand sidebar (or at the bottom of the page, depending on your device).
Research Highlights
- We developed the currently most robust time-dependent Density Functional Theory (TD-DFT) methods for organic molecules; so called long-range corrected double hybrid with long-range correction (range-separation). You can find the paper here. Preceding developments, namely the first range-separated double hybrids for excited states, were published here with a follow-up paper on triplet excited states published here. A study confirming that range separation is needed for charge-transfer excitations even when using double hybrids can be found here. All our methods are available for free in ORCA5.
- Read our new account on DFT applications specifically written for non-expert users and people new in the field. It can be accessed for free here. A similar account on TD-DFT for excited states written for non-expert users can be accessed for free here.
- Dr Lars Goerigk won the 2022 Pople Medal by the Asia-Pacific Association of Theoretical & Computational Chemists, the highest award for a theoretical/computational chemist under the age of 45 in the Asia Pacific.
- Dr Lars Goerigk won a 2020 Rennie Memorial Medal, the highest national early-career prize awarded by the Royal Australian Chemical Institute. For other RACI 2020 awardees, click here.
-
Watch Dr Lars Goerigk talk about our research and his 2019 Le Fèvre Medal awarded by the Australian Academy of Science for his contributions to the field of DFT. To see the citations for all 20 of the Academy of Science’s 2019 Honorific Awards, click here.
- A selection of our most recent DFT benchmarking contributions for methods users and developers:
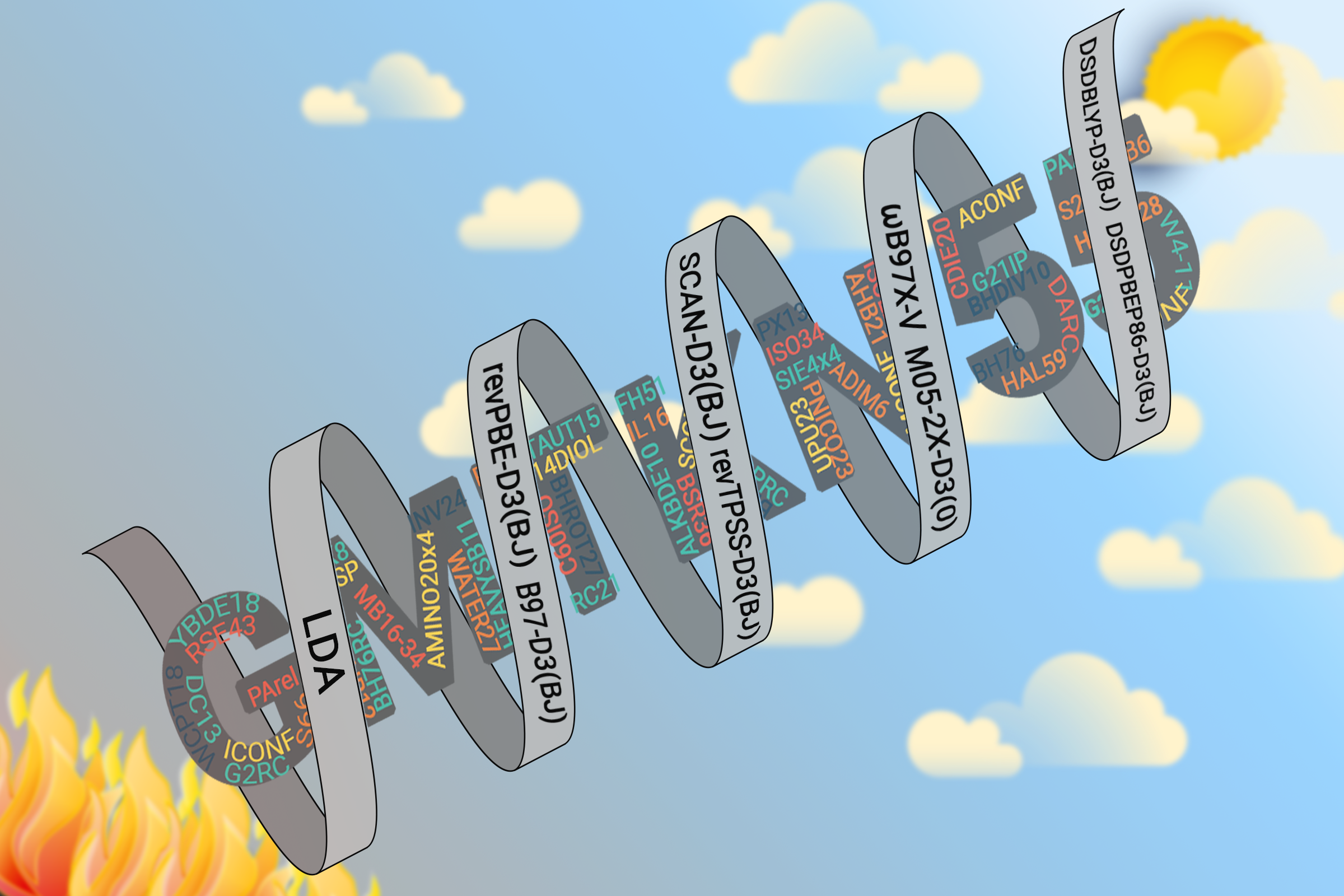

- Top left picture: GMTKN55: a database for general main-group thermochemistry, kinetics and noncovoalent interactions. Find the open-access First GMTKN55 paper here. Second and third GMTKN55 papers are here and here. Tips for students and new DFT users with a GMTKN55-based comment on popular approaches can be found here. Our current recommendations for DFT (as of 2 September 2020) can be found here. Access the GMTKN55 website here.
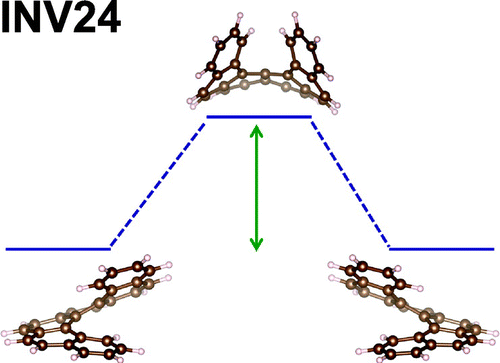
- Top right picture: INV24: The first test set for inversion barriers available as Open Access.
- Bottom left picture: CHAL336: The most comprehensive test set for chalcogen-bonding interactions can be found here.
- Bottom right picture: MME55: The first, accurate test set for metalloenzyme reactions can be found here.
- We also published guidelines for generating accurate reference data for models representing enzymatically catalyzed reactions incl. a benchmark set and DFT recommendations. The relevant papers are here and here.
See more on our research here. Our publications are listed here.
Latest News
18 June 2025
It’s been a while since a group update has been given, so here are the highlights:
- Ben and Chloe joined the group as Masters students in March and have already made great progress. Chloe will also conduct experiments, as she is co-supervised by Prof. Evan Bieske
- Congratulations, Amy, for submitting your PhD thesis
- Congratulations, Erica, for submitting your Masters thesis. Erica will be leaving the group next week. You’ll be missed, Erica. All the best for your future career.
- Quyen’s 12 months stay here is coming to an end. She will return to Belgium soon to complete her joint-PhD degree with KU Leuven there.
- Lars Goerigk has joined the Editorial Advisory Board of The Journal of Computational Chemistry.